
Our research focuses on discovering and developing new chemical transformations that enable the construction of biologically active molecular entities and the valorization of natural products and abundant biomass‑derived feedstocks.
To support and guide our synthetic efforts, we employ Density Functional Theory to elucidate reaction mechanisms and gain deeper insight into photochemical processes. These computational studies enhance our understanding of molecular behavior relevant to applications in materials science and medical chemistry
ReseaRch Topics

Valorization of Quinic Acid
Quinic acid, a naturally occurring compound found in coffee beans, fruits, and even food waste, is more than just a plant metabolite. With its rich array of hydroxyl groups arranged around a rigid cyclic backbone, quinic acid offers a unique platform for building complex, chiral molecules. These features make it an ideal starting point for crafting biologically active structures such as cyclitols and carbasugars, which are of growing interest in medicinal chemistry and drug discovery.
Our research explores how to unlock the full synthetic potential of quinic acid using cutting-edge chemical techniques. By leveraging modern catalytic methods, spectroscopic tools, and computational modeling, we aim to transform this abundant natural product into a versatile toolkit for molecular design. Whether it's through selective functionalization, protecting-group-free strategies, or photoredox catalysis, each transformation adds a new layer of complexity and utility to this deceptively simple molecule.
In doing so, we not only expand the chemical space accessible from renewable resources but also contribute to a more sustainable and innovative future for organic synthesis.
Selected publications:
Tetrahedron Green Chem., 2025, 5, 100070. (DOI: 10.1016/j.tgchem.2025.100070)
Org. Lett. 2021, 23, 3083. (DOI: 10.1021/acs.orglett.1c00755)
Org. Lett. 2020, 22, 8370-8375. (DOI: 10.1021/acs.orglett.0c02995)
Kindly supported by: FCT PhD scholarships and Project AQ+i (PTDC/QUI-QOR/1131/2020) and Academy of Finland (grants: 4248724, 294067 and 319198)

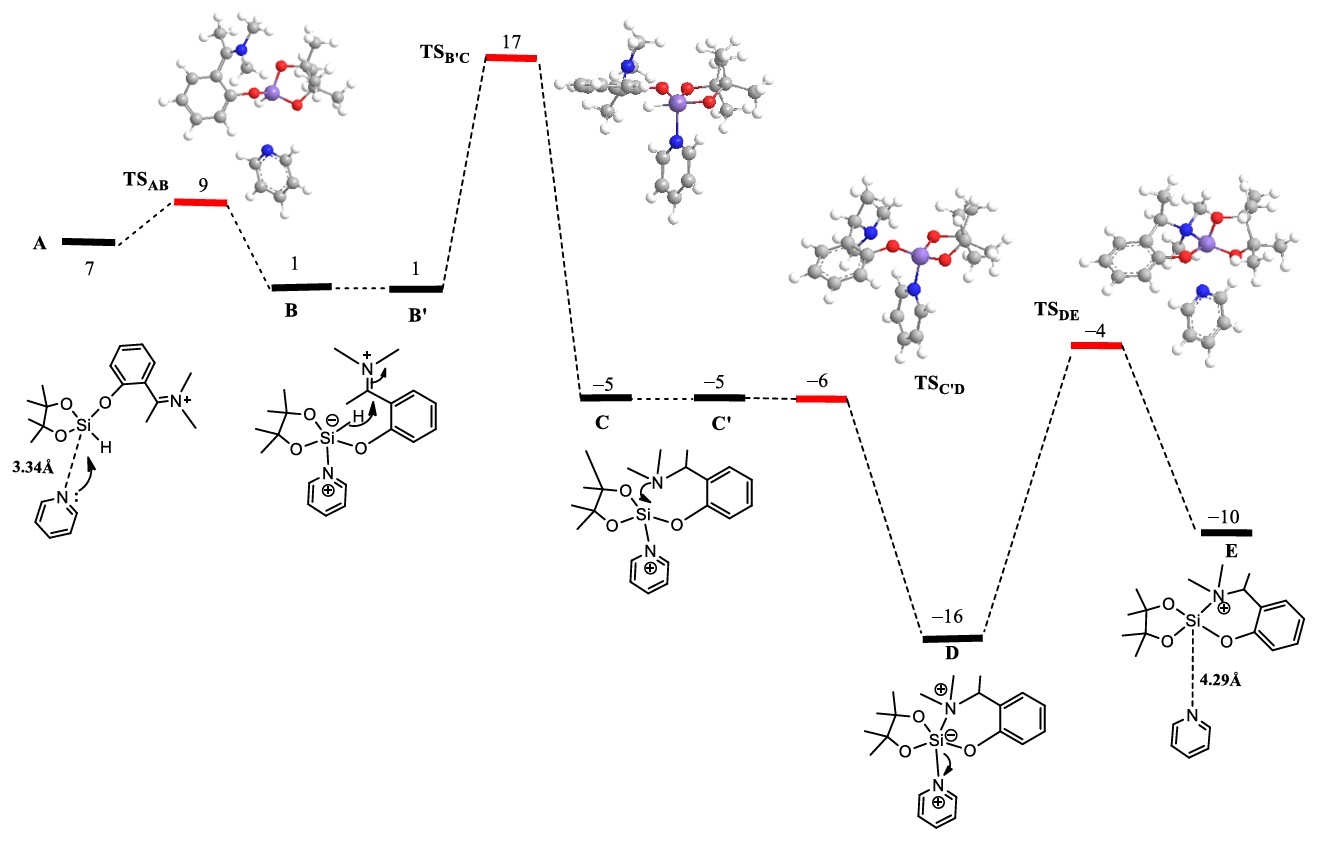
Hydrosilylations
In synthetic chemistry, reduction reactions are commonly used to modify molecular structures. Traditionally, this meant relying on harsh reagents like metal hydrides or high-pressure hydrogenation—effective, but not always elegant. Catalytic hydrosilylation is a modern, metal-free alternative that’s turning heads for its precision, versatility, and environmental friendliness.
We have developed a novel chlorohydrosilane derived from pinacol—a compact, stable molecule that, when activated by a Lewis base, becomes a powerful tool for reducing carbon-heteroatom double bonds. From selective reductions of salicylaldehydes to the streamlined synthesis of aminoalkylphenols, we’re seeing transformations that were once cumbersome become clean, efficient, and surprisingly gentle.
Computational studies reveal how subtle changes in molecular geometry and electronic structure influence reactivity, providing insight into the design of efficient and selective reduction systems.
Selected publications:
Org. Lett. 2019, 21, 1402-1406. (DOI: 10.1021/acs.orglett.9b00121)
Eur. J. Org. Chem. 2018, 2910-2917. (DOI: 10.1002/ejoc.201800544)
Kindly supported by: FCT project Spartalysis (PTDC/QUI-QOR/1786/2021) and Academy of Finland (grants: 4248724, 294067 and 319198)

Aminoalkylphenols
The multicomponent Petasis borono-Mannich reaction is an excellent tool for the preparation of alkylaminophenols, given its easy operability and the possibility for structural diversification of the products. These small molecules have inspired us in the development of new antibacterial and antitumor agents. We keep searching for other pharmaceutical uses of these compounds either by looking into unraveled biological properties or by developing technological applications for biological systems.
Selected publications:
RSC Med. Chem., 2025, 16, 6204-6213 (DOI: 10.1039/D5MD00585J)
J. Med. Chem. 2021, 64, 10908. (DOI: 10.1021/acs.jmedchem.1c00277)
Eur. J. Med. Chem. 2021, 220, 113459. (DOI: 10.1016/j.ejmech.2021.113459)
Br. J. Pharmacol., 2024, 181, 107. (DOI: 10.1111/bph.16141 )
Kindly supported by: \ Academy of Finland (grants: 4248724, 294067 and 319198) and Jane and Aatos Erkko Foundation

OrganoSilicon Compounds
Organosilicon chemistry offers unusual opportunities to control highly reactive intermediates, and reagents such as acylsilanes and bis(silyl)peroxides illustrate this particularly well. Acylsilanes act as reliable carbene precursors, and we have used visible light to activate them through 1,2‑Brook rearrangements. This has allowed us to develop an intramolecular [2+2] photocycloaddition that builds diaryl‑substituted silacycles directly from simple starting materials. We also established a metal‑free formal [4+1] cycloaddition that uses photogenerated siloxycarbenes to access structurally rich, functionalized cyclopentenes.
Our curiosity has been extended to bis(silyl)peroxides, which can offer a controlled route to carbon‑centered radicals. Their enhanced stability—compared with traditional peroxide reagents—provides a safer and more predictable platform for radical chemistry. Through this approach, we aim to expand the set of radical transformations available under mild, operationally simple conditions.
Together, these silicon‑based platforms demonstrate how tuning electronic effects at silicon can enable clean entry into reactive carbene and radical manifolds. By providing controlled access to reactive species, they open practical pathways for constructing complex molecular frameworks.
Selected publications:
J. Org. Chem, 2022, 87, 8910. (DOI: 10.1021/acs.joc.2c00591)
Org. Chem. Frontiers, 2019, 6, 3793-3798. (DOI: 10.1039/c9qo01028a)
Kindly supported by: FCT PhD scholarships and BiSiPe4Tame project (2023.17015.ICDT)
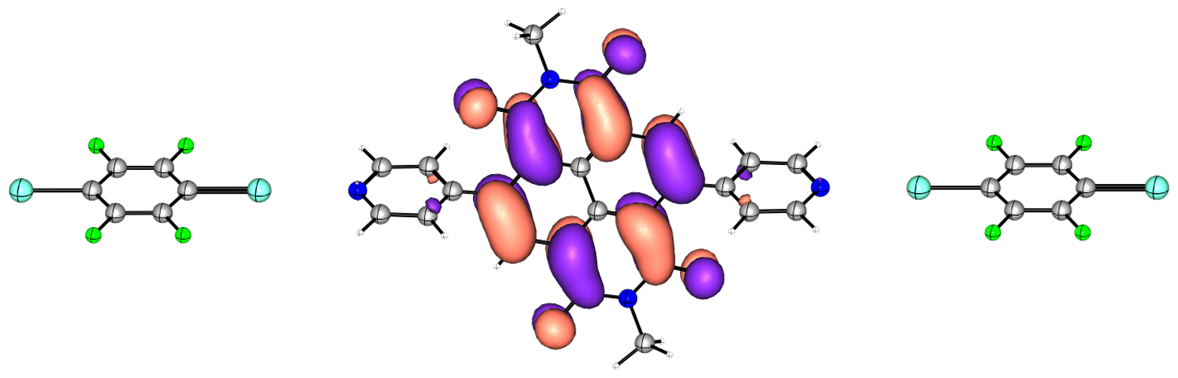
Density Functional Theory
DFT calculations play a central role in our group’s work. Across our projects - from metal-free reductions and carbene or radical generation to heterocycle assembly and semiconductor design - we use DFT to elucidate reaction mechanisms, identify key intermediates, and rationalize selectivity trends.
Beyond supporting our own synthetic studies, we collaborate with experimental groups to model diverse material properties, including electronic structure, molecular packing, charge‑transport behaviour, and excited‑state processes. These computational insights strengthen the design of new reagents, functional molecules, and organic electronic materials.
Selected publications:
Angew. Chem. Int. Ed., 2025, 64, e202424979. (DOI: 10.1002/anie.202424979)
Org. Lett., 2025, 27, 11757. (DOI: 10.1021/acs.orglett.5c03509)
ACS Catal., 2023, 13, 1916. (DOI: 10.1021/acscatal.2c04867)
Mater. Adv., 2022, 3, 1703. (DOI: 10.1039/D1MA00438G)