Welcome to our group website
Main interests
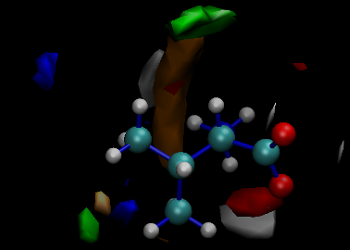
Reactions at Interfaces: Several different computational techniques (quantum mechanical (QM), molecular mechanics (MM), QM/MM, molecular dynamics (MD) or coarse-grained (CG) simulations, docking, 3D-QSAR, microkinetic modelling, ...) are being used to analyze the interactions and reactions of adsorbates (in gaseous or in solution) with materials or enzymes. The calculations are being used to interpret experimental results obtained for heterogeneous catalysis at pure metallic, bimetallic or metal supported surfaces, to design new compounds with capability to inhibit falcipains and to understand the templated condensation of silica in the synthesis of periodic mesoporous (organo)silicas (PMO or PMS materials).
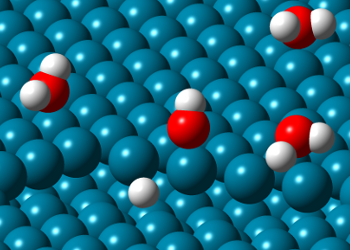
Adsorption of Gases in Porous Materials: Predictions of equilibrium adsorption and permeabilities for pure- and multi-components on porous materials (some synthesized at CICECO, e.g. PMO materials) are being obtained from grand canonical Monte Carlo (GCMC) and molecular dynamics (MD) simulations. The isotherms are compared with experimentally measured curves in order to improve the models. The idea is to find/optimize materials for gas separation (propane/propylene; CO2/CH4; ...). Density functional theory (DFT) is also being employed to analyze specific gas-substrate interactions.
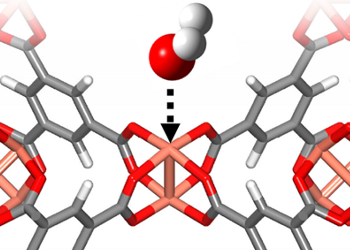
Interactions of Ionic Liquids with (Bio)organics: The liquid-liquid equilibrium and solubility behaviors of mixtures of ionic liquids and (bio)organic molecules are being analyzed by density functional theory (DFT) and by molecular dynamics (MD) simulations. These studies are being conducted in collaboration with experimental work (nuclear magnetic resonance shifts, density measurements, excess volumes, etc.) performed at CICECO. The computational techniques are being employed to analyze which are the preferred interactions between the constituting species.