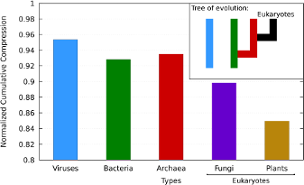
 The Kolmogorov complexity furnishes several ways for studying different natural processes that can be expressed using sequences of symbols from a finite alphabet, such as the case of DNA sequences. Although the Kolmogorov complexity is not algorithmically computable, it can be approximated by lossless normal compressors. In this paper, we use a specific DNA compressor to approximate the Kolmogorov complexity and we assess it regarding its normality. Then, we use it on several datasets, that are constituted by different DNA sequences, representing complete genomes of different species and domains. We show several evolution-related insights associated with the complexity, namely that, globally, archaea have higher relative complexity than bacteria and eukaryotes.
The Kolmogorov complexity furnishes several ways for studying different natural processes that can be expressed using sequences of symbols from a finite alphabet, such as the case of DNA sequences. Although the Kolmogorov complexity is not algorithmically computable, it can be approximated by lossless normal compressors. In this paper, we use a specific DNA compressor to approximate the Kolmogorov complexity and we assess it regarding its normality. Then, we use it on several datasets, that are constituted by different DNA sequences, representing complete genomes of different species and domains. We show several evolution-related insights associated with the complexity, namely that, globally, archaea have higher relative complexity than bacteria and eukaryotes.
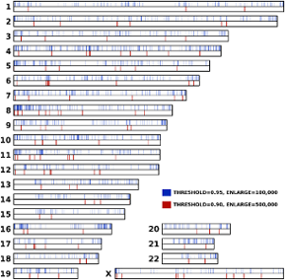 Species-specific DNA regions are segments that are unique or share high dissimilarity relatively to close species. Their discovery is important, because they allow the localization of evolutionary traits that are often related to novel functionalities and, sometimes, diseases.
We have detected distinct DNA regions specific in the modern human, when compared to a Neanderthal high-quality genome sequence obtained from a bone of a Siberian woman. The bone is around 50,000 years old and the DNA raw data totalizes more than 418 GB. Since the data size required for localizing efficiently such events is very high, it is not practical to store the model on a table or hash table. Thus, we propose a probabilistic method to map and visualize those regions. The time complexity of the method is linear.
Species-specific DNA regions are segments that are unique or share high dissimilarity relatively to close species. Their discovery is important, because they allow the localization of evolutionary traits that are often related to novel functionalities and, sometimes, diseases.
We have detected distinct DNA regions specific in the modern human, when compared to a Neanderthal high-quality genome sequence obtained from a bone of a Siberian woman. The bone is around 50,000 years old and the DNA raw data totalizes more than 418 GB. Since the data size required for localizing efficiently such events is very high, it is not practical to store the model on a table or hash table. Thus, we propose a probabilistic method to map and visualize those regions. The time complexity of the method is linear.
The computational tool is available at http://pratas.github.io/chester.
The results, computed in approximately two days using a single CPU core, show several regions with documented neanderthal absent regions, namely genes associated with the brain (neurotransmiters and synapses), hearing, blood, fertility and the immune system. However, it also shows several undocumented regions, that may express new functions linked with the evolution of the modern human.
 NGS (next-generation sequencing) is bringing the need to efficiently handle large volumes of patient data, maintaining privacy laws, such as those with secure protocols that ensure patients DNA confidentiality. Although there are multiple file representations for genomic data, the FASTA format is perhaps the most used and popular. As far as we know, FASTA encryption is being addressed with general purpose encryption methods, without exploring a compact representation. In this paper, we propose Cryfa, a new fast encryption method to store securely FASTA files in a compact form. The main differences between a general encryption approach and Cryfa are the reduction of storage, up to approximately three times, without compromising security, and the possibility of integration with pipelines. The core of the encryption method uses a symmetric approach, the AES (Advanced Encryption Standard). Cryfa implementation is freely available, under license GPLv3, at https://github.com/pratas/cryfa.
NGS (next-generation sequencing) is bringing the need to efficiently handle large volumes of patient data, maintaining privacy laws, such as those with secure protocols that ensure patients DNA confidentiality. Although there are multiple file representations for genomic data, the FASTA format is perhaps the most used and popular. As far as we know, FASTA encryption is being addressed with general purpose encryption methods, without exploring a compact representation. In this paper, we propose Cryfa, a new fast encryption method to store securely FASTA files in a compact form. The main differences between a general encryption approach and Cryfa are the reduction of storage, up to approximately three times, without compromising security, and the possibility of integration with pipelines. The core of the encryption method uses a symmetric approach, the AES (Advanced Encryption Standard). Cryfa implementation is freely available, under license GPLv3, at https://github.com/pratas/cryfa.
 Referential compression is one of the fundamental operations for storing and analyzing DNA data. The models that incorporate relative compression, a special case of referential compression, are being steadily improved, namely those which are based on Markov models. In this paper, we propose a new model, the substitutional tolerant Markov model (STMM), which can be used in cooperation with regular Markov models to improve compression efficiency. We assessed its impact on synthetic and real DNA sequences, showing a substantial improvement in compression, while only slightly increasing the computation time. In particular, it shows high efficiency in modeling species that have split less than 40 million years ago.
Referential compression is one of the fundamental operations for storing and analyzing DNA data. The models that incorporate relative compression, a special case of referential compression, are being steadily improved, namely those which are based on Markov models. In this paper, we propose a new model, the substitutional tolerant Markov model (STMM), which can be used in cooperation with regular Markov models to improve compression efficiency. We assessed its impact on synthetic and real DNA sequences, showing a substantial improvement in compression, while only slightly increasing the computation time. In particular, it shows high efficiency in modeling species that have split less than 40 million years ago.
 In this paper, we propose a computational approach to quantify inverted repeats. This is important, because it is known that the presence of inverted repeats in genomic data may be associated to certain chromosomal rearrangements. First, we present a reference-based relative compression method, which employs statistical characteristics of the genomic data. Then, for determining the similarity between genomic sequences, we use the normalized relative compression measure, which is light-weight regarding computational time and memory. Testing this approach on various species, including human, chimpanzee, gorilla, chicken, turkey and archaea genomes, we unveil unreported results that may support several evolution insights.
In this paper, we propose a computational approach to quantify inverted repeats. This is important, because it is known that the presence of inverted repeats in genomic data may be associated to certain chromosomal rearrangements. First, we present a reference-based relative compression method, which employs statistical characteristics of the genomic data. Then, for determining the similarity between genomic sequences, we use the normalized relative compression measure, which is light-weight regarding computational time and memory. Testing this approach on various species, including human, chimpanzee, gorilla, chicken, turkey and archaea genomes, we unveil unreported results that may support several evolution insights.
 The ever increasing growth of the production of high-throughput sequencing data poses a serious challenge to the storage, processing and transmission of these data. As frequently stated, it is a data deluge. Compression is essential to address this challenge—it reduces storage space and processing costs, along with speeding up data transmission. In this paper, we provide a comprehensive survey of existing compression approaches, that are specialized for biological data, including protein and DNA sequences. Also, we devote an important part of the paper to the approaches proposed for the compression of different file formats, such as FASTA, as well as FASTQ and SAM/BAM, which contain quality scores and metadata, in addition to the biological sequences. Then, we present a comparison of the performance of several methods, in terms of compression ratio, memory usage and compression/decompression time. Finally, we present some suggestions for future research on biological data compression.
The ever increasing growth of the production of high-throughput sequencing data poses a serious challenge to the storage, processing and transmission of these data. As frequently stated, it is a data deluge. Compression is essential to address this challenge—it reduces storage space and processing costs, along with speeding up data transmission. In this paper, we provide a comprehensive survey of existing compression approaches, that are specialized for biological data, including protein and DNA sequences. Also, we devote an important part of the paper to the approaches proposed for the compression of different file formats, such as FASTA, as well as FASTQ and SAM/BAM, which contain quality scores and metadata, in addition to the biological sequences. Then, we present a comparison of the performance of several methods, in terms of compression ratio, memory usage and compression/decompression time. Finally, we present some suggestions for future research on biological data compression.
 The number of genomic sequences is growing substantially. Besides discarding part of the data, the only efficient possibility for coping with this trend is data compression. We present an efficient compressor for genomic sequences, allowing both reference-free and referential
compression. This compressor uses a mixture of context models of several orders, according to two model classes: reference and target. A new type of context model, which is capable of tolerating substitution errors, is introduced. For ensuring flexibility regarding hardware specifications, the compressor uses cache-hashes in high order models. The results show additional compression gains over several specific top tools in different levels of redundancy. The implementation is available at https://github.com/pratas/geco.
The number of genomic sequences is growing substantially. Besides discarding part of the data, the only efficient possibility for coping with this trend is data compression. We present an efficient compressor for genomic sequences, allowing both reference-free and referential
compression. This compressor uses a mixture of context models of several orders, according to two model classes: reference and target. A new type of context model, which is capable of tolerating substitution errors, is introduced. For ensuring flexibility regarding hardware specifications, the compressor uses cache-hashes in high order models. The results show additional compression gains over several specific top tools in different levels of redundancy. The implementation is available at https://github.com/pratas/geco.
Authorship attribution is a classical classification problem. We use it here to illustrate the performance of a compression-based measure that relies on the notion of relative compression. Besides comparing with recent approaches that use multiple discriminant analysis
and support vector machines, we compare it with the Normalized Conditional Compression Distance (a direct approximation of the Normalized Information Distance) and the popular Normalized Compression Distance. The Normalized Relative Compression (NRC) attained 100% correct classification in the data set used, showing consistency between the compression ratio and the classification performance, a characteri
stic not always present in other compression-based measures.
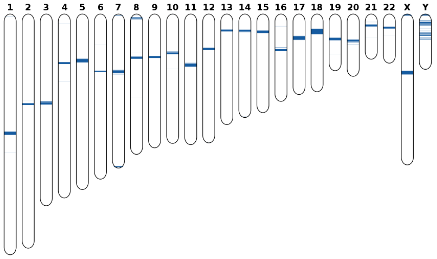 Human specific regions are DNA segments that are unique or share high dissimilarity rates relatively to close species, namely primates. Their existence is important to localize evolutionary traits that are often related to novel functionality, besides its obvious discriminative ability. We propose an unsupervised method, and an associated tool, to detect and visualise these regions. It is based on the detection of relative absent words (RAWs), using a probabilistic high depth model. The experimental results show several regions that are associated with documented human specific regions, namely centromeres and several genes, such as those related with olfact. However, it also shows several undocumented ones, that may express trends in human evolution. Availability and Implementation: CHESTER is freely available for
non-commercial purposes at https://github.com/pratas/chester.
Human specific regions are DNA segments that are unique or share high dissimilarity rates relatively to close species, namely primates. Their existence is important to localize evolutionary traits that are often related to novel functionality, besides its obvious discriminative ability. We propose an unsupervised method, and an associated tool, to detect and visualise these regions. It is based on the detection of relative absent words (RAWs), using a probabilistic high depth model. The experimental results show several regions that are associated with documented human specific regions, namely centromeres and several genes, such as those related with olfact. However, it also shows several undocumented ones, that may express trends in human evolution. Availability and Implementation: CHESTER is freely available for
non-commercial purposes at https://github.com/pratas/chester.
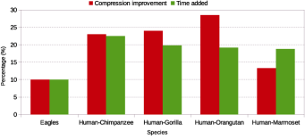
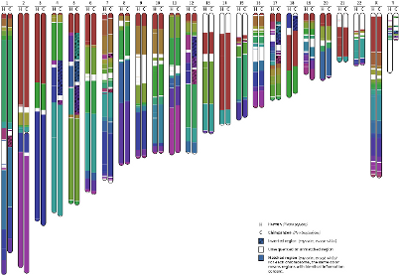 Species evolution is indirectly registered in their genomic structure. The emergence and advances in sequencing technology provided a way to access genome information, namely to identify and study evolutionary macro-events, as well as chromosome alterations for clinical purposes. This paper describes a completely alignment-free computational method, based on a blind unsupervised approach, to detect large-scale and small-scale genomic rearrangements between pairs of DNA sequences. To illustrate the power and usefulness of the method we give complete chromosomal information maps for the pairs human-chimpanzee and human-orangutan. The tool by means of which these results were obtained has been made publicly available and is described in detail.
Species evolution is indirectly registered in their genomic structure. The emergence and advances in sequencing technology provided a way to access genome information, namely to identify and study evolutionary macro-events, as well as chromosome alterations for clinical purposes. This paper describes a completely alignment-free computational method, based on a blind unsupervised approach, to detect large-scale and small-scale genomic rearrangements between pairs of DNA sequences. To illustrate the power and usefulness of the method we give complete chromosomal information maps for the pairs human-chimpanzee and human-orangutan. The tool by means of which these results were obtained has been made publicly available and is described in detail.
Availability and Implementation: SMASH is freely available for
non-commercial purposes at https://github.com/pratas/smash
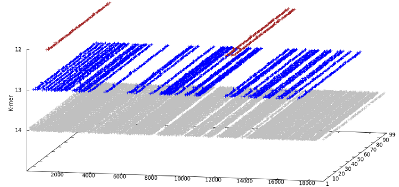 Ebola virus causes high mortality hemorrhagic
fevers, with more than 25k cases and 10k deaths in
the current outbreak. Only experimental therapies are
available, thus, novel diagnosis tools and druggable targets
are needed. Analysis of Ebola virus genomes from
the current outbreak reveals the presence of short DNA
sequences that appear nowhere in the human genome. We identify
the shortest such sequences with lengths between 12 and 14.
Only three absent sequences of length 12 exist and they
consistently appear at the same location on two of the Ebola
virus proteins, in all Ebola virus genomes, but nowhere in the
human genome. The alignment-free method used is able to
identify pathogen-specific signatures for quick and precise
action against infectious agents, of which the current Ebola
virus outbreak provides a compelling example. EAGLE is freely available, under GPLv3, at https://github.com/pratas/eagle.
Ebola virus causes high mortality hemorrhagic
fevers, with more than 25k cases and 10k deaths in
the current outbreak. Only experimental therapies are
available, thus, novel diagnosis tools and druggable targets
are needed. Analysis of Ebola virus genomes from
the current outbreak reveals the presence of short DNA
sequences that appear nowhere in the human genome. We identify
the shortest such sequences with lengths between 12 and 14.
Only three absent sequences of length 12 exist and they
consistently appear at the same location on two of the Ebola
virus proteins, in all Ebola virus genomes, but nowhere in the
human genome. The alignment-free method used is able to
identify pathogen-specific signatures for quick and precise
action against infectious agents, of which the current Ebola
virus outbreak provides a compelling example. EAGLE is freely available, under GPLv3, at https://github.com/pratas/eagle.
 In the last decade, the cost of genomic sequencing has been decreasing so much that researchers all over the world accumulate huge amounts of data for present and future use. These genomic data need to be efficiently stored, because storage cost is not decreasing as fast as the cost of sequencing. In order to overcome this problem, the most popular general-purpose compression tool, gzip, is usually used. However, these tools were not specifically designed to compress this kind of data, and often fall short when the intention is to reduce the data size as much as possible. There are several compression algorithms available, even for genomic data, but very few have been designed to deal with Whole Genome Alignments, containing alignments between entire genomes of several species. In this paper, we present a lossless compression tool, MAFCO, specifically designed to compress MAF (Multiple Alignment Format) files. Compared to gzip, the proposed tool attains a compression gain from 34% to 57%, depending on the data set. When compared to a recent dedicated method, which is not compatible with some data sets, the compression gain of MAFCO is about 9%. Both source-code and binaries for several operating systems are freely available for non-commercial use at:
http://bioinformatics.ua.pt/software/mafco.
In the last decade, the cost of genomic sequencing has been decreasing so much that researchers all over the world accumulate huge amounts of data for present and future use. These genomic data need to be efficiently stored, because storage cost is not decreasing as fast as the cost of sequencing. In order to overcome this problem, the most popular general-purpose compression tool, gzip, is usually used. However, these tools were not specifically designed to compress this kind of data, and often fall short when the intention is to reduce the data size as much as possible. There are several compression algorithms available, even for genomic data, but very few have been designed to deal with Whole Genome Alignments, containing alignments between entire genomes of several species. In this paper, we present a lossless compression tool, MAFCO, specifically designed to compress MAF (Multiple Alignment Format) files. Compared to gzip, the proposed tool attains a compression gain from 34% to 57%, depending on the data set. When compared to a recent dedicated method, which is not compatible with some data sets, the compression gain of MAFCO is about 9%. Both source-code and binaries for several operating systems are freely available for non-commercial use at:
http://bioinformatics.ua.pt/software/mafco.
 The emerging next-generation sequencing (NGS) is bringing, besides the natural huge amounts of data, an avalanche of new specialized tools (for analysis, compression, alignment, among others) and large public and private network infrastructures. Therefore, a direct necessity of specific simulation tools for testing and benchmarking is rising, such as a flexible and portable FASTQ read simulator, without the need of a reference sequence, yet correctly prepared for producing approximately the same characteristics as real data.
We present XS, a skilled FASTQ read simulation tool, flexible, portable (does not need a reference sequence) and tunable in terms of sequence complexity. It has several running modes, depending on the time and memory available, and is aimed at testing computing infrastructures, namely cloud computing of large-scale projects, and testing FASTQ compression algorithms. Moreover, XS offers the possibility of simulating the three main FASTQ components individually (headers, DNA sequences and quality-scores).
XS provides an efficient and convenient method for fast simulation of FASTQ files, such as those from Ion Torrent (currently uncovered by other simulators), Roche-454, Illumina and ABI-SOLiD sequencing machines. This tool is publicly available at http://bioinformatics.ua.pt/software/xs/.
The emerging next-generation sequencing (NGS) is bringing, besides the natural huge amounts of data, an avalanche of new specialized tools (for analysis, compression, alignment, among others) and large public and private network infrastructures. Therefore, a direct necessity of specific simulation tools for testing and benchmarking is rising, such as a flexible and portable FASTQ read simulator, without the need of a reference sequence, yet correctly prepared for producing approximately the same characteristics as real data.
We present XS, a skilled FASTQ read simulation tool, flexible, portable (does not need a reference sequence) and tunable in terms of sequence complexity. It has several running modes, depending on the time and memory available, and is aimed at testing computing infrastructures, namely cloud computing of large-scale projects, and testing FASTQ compression algorithms. Moreover, XS offers the possibility of simulating the three main FASTQ components individually (headers, DNA sequences and quality-scores).
XS provides an efficient and convenient method for fast simulation of FASTQ files, such as those from Ion Torrent (currently uncovered by other simulators), Roche-454, Illumina and ABI-SOLiD sequencing machines. This tool is publicly available at http://bioinformatics.ua.pt/software/xs/.
 The data deluge phenomenon is becoming a serious problem in most genomic centers. To alleviate it, general purpose tools, such as gzip, are used to compress the data. However, although pervasive and easy to use, these tools fall short when the intention is to reduce as much as possible the data, for example, for medium- and long-term storage. A number of algorithms have been proposed for the compression of genomics data, but unfortunately only a few of them have been made available as usable and reliable compression tools.
In this article, we describe one such tool, MFCompress, specially designed for the compression of FASTA and multi-FASTA files. In comparison to gzip and applied to multi-FASTA files, MFCompress can provide additional average compression gains of almost 50%, i.e. it potentially doubles the available storage, although at the cost of some more computation time. On highly redundant datasets, and in comparison with gzip, 8-fold size reductions have been obtained.
Both source code and binaries for several operating systems are freely available for non-commercial use at http://bioinformatics.ua.pt/software/mfcompress.
The data deluge phenomenon is becoming a serious problem in most genomic centers. To alleviate it, general purpose tools, such as gzip, are used to compress the data. However, although pervasive and easy to use, these tools fall short when the intention is to reduce as much as possible the data, for example, for medium- and long-term storage. A number of algorithms have been proposed for the compression of genomics data, but unfortunately only a few of them have been made available as usable and reliable compression tools.
In this article, we describe one such tool, MFCompress, specially designed for the compression of FASTA and multi-FASTA files. In comparison to gzip and applied to multi-FASTA files, MFCompress can provide additional average compression gains of almost 50%, i.e. it potentially doubles the available storage, although at the cost of some more computation time. On highly redundant datasets, and in comparison with gzip, 8-fold size reductions have been obtained.
Both source code and binaries for several operating systems are freely available for non-commercial use at http://bioinformatics.ua.pt/software/mfcompress.
Data summarization and triage is one of the current top challenges in visual analytics. The goal is to let users visually inspect large data sets and examine or request data with particular characteristics. The need for summarization and visual analytics is also felt when dealing with digital representations of DNA sequences. Genomic data sets are growing rapidly, making their analysis increasingly more difficult, and raising the need for new, scalable tools. For example, being able to look at very large DNA sequences while immediately identifying potentially interesting regions would provide the biologist with a flexible exploratory and analytical tool. In this paper we present a new concept, the “information profile”, which provides a quantitative measure of the local complexity of a DNA sequence, independently of the direction of processing. The computation of the information profiles is computationally tractable: we show that it can be done in time proportional to the length of the sequence. We also describe a tool to compute the information profiles of a given DNA sequence, and use the genome of the fission yeast Schizosaccharomyces pombe strain 972 h− and five human chromosomes 22 for illustration. We show that information profiles are useful for detecting large-scale genomic regularities by visual inspection. Several discovery strategies are possible, including the standalone analysis of single sequences, the comparative analysis of sequences from individuals from the same species, and the comparative analysis of sequences from different organisms. The comparison scale can be varied, allowing the users to zoom-in on specific details, or obtain a broad overview of a long segment. Software applications have been made available for non-commercial use at http://bioinformatics.ua.pt/software/dna-at-glance.
 A particularly voluminous dataset in molecular genomics, known as whole genome alignments, has gained considerable importance over the last years. In this paper, we propose a compression modeling approach for the multiple sequence alignment (MSA) blocks, which make up most of these datasets. Our method is based on a mixture of finite-context models. Contrarily to other recent approaches, it addresses both the DNA bases and gap symbols at once, better exploring the existing correlations. For comparison with previous methods, our algorithm was tested in the multiz28way dataset. On average, it attained 0.94 bits per symbol, approximately 7% better than the previous best, for a similar computational complexity. We also tested the model in the most recent dataset, multiz46way. In this dataset, that contains alignments of 46 different species, our compression model achieved an average of 0.72 bits per MSA block symbol.
A particularly voluminous dataset in molecular genomics, known as whole genome alignments, has gained considerable importance over the last years. In this paper, we propose a compression modeling approach for the multiple sequence alignment (MSA) blocks, which make up most of these datasets. Our method is based on a mixture of finite-context models. Contrarily to other recent approaches, it addresses both the DNA bases and gap symbols at once, better exploring the existing correlations. For comparison with previous methods, our algorithm was tested in the multiz28way dataset. On average, it attained 0.94 bits per symbol, approximately 7% better than the previous best, for a similar computational complexity. We also tested the model in the most recent dataset, multiz46way. In this dataset, that contains alignments of 46 different species, our compression model achieved an average of 0.72 bits per MSA block symbol.
 Genome assemblies are typically compared with respect to their contiguity, coverage, and accuracy. We propose a genome-wide, alignment-free genomic distance based on compressed maximal exact matches and suggest adding it to the benchmark of commonly used assembly quality metrics. Maximal exact matches are perfect repeats, without gaps or misspellings, which cannot be further extended to either their left- or right-end side without loss of similarity. The genomic distance here proposed is based on the normalized compression distance, an information-theoretic measure of the relative compressibility of two sequences estimated using multiple finite-context models. This measure exposes similarities between the sequences, as well as, the nesting structure underlying the assembly of larger maximal exact matches from smaller ones. We use four human genome assemblies for illustration and discuss the impact of genome sequencing and assembly in the final content of maximal exact matches and the genomic distance here proposed.
Genome assemblies are typically compared with respect to their contiguity, coverage, and accuracy. We propose a genome-wide, alignment-free genomic distance based on compressed maximal exact matches and suggest adding it to the benchmark of commonly used assembly quality metrics. Maximal exact matches are perfect repeats, without gaps or misspellings, which cannot be further extended to either their left- or right-end side without loss of similarity. The genomic distance here proposed is based on the normalized compression distance, an information-theoretic measure of the relative compressibility of two sequences estimated using multiple finite-context models. This measure exposes similarities between the sequences, as well as, the nesting structure underlying the assembly of larger maximal exact matches from smaller ones. We use four human genome assemblies for illustration and discuss the impact of genome sequencing and assembly in the final content of maximal exact matches and the genomic distance here proposed.
 Research in the genomic sciences is confronted with the volume of sequencing and resequencing data increasing at a higher pace than that of data storage and communication resources, shifting a significant part of research budgets from the sequencing component of a project to the computational one. Hence, being able to efficiently store sequencing and resequencing data is a problem of paramount importance. In this article, we describe GReEn (Genome Resequencing Encoding), a tool for compressing genome resequencing data using a reference genome sequence. It overcomes some drawbacks of the recently proposed tool GRS, namely, the possibility of compressing sequences that cannot be handled by GRS, faster running times and compression gains of over 100-fold for some sequences. This tool is freely available for non-commercial use at http://bioinformatics.ua.pt/software/green.
Research in the genomic sciences is confronted with the volume of sequencing and resequencing data increasing at a higher pace than that of data storage and communication resources, shifting a significant part of research budgets from the sequencing component of a project to the computational one. Hence, being able to efficiently store sequencing and resequencing data is a problem of paramount importance. In this article, we describe GReEn (Genome Resequencing Encoding), a tool for compressing genome resequencing data using a reference genome sequence. It overcomes some drawbacks of the recently proposed tool GRS, namely, the possibility of compressing sequences that cannot be handled by GRS, faster running times and compression gains of over 100-fold for some sequences. This tool is freely available for non-commercial use at http://bioinformatics.ua.pt/software/green.
Chapters:
- Armando J. Pinho, Diogo Pratas, Sara Pinto Garcia. Compressing resequencing data with GReEn. Deep Sequencing Data Analysis, Noam Shomron (Ed.), Humana Press, vol. 1038 (Methods in Molecular Biology), July 2013. doi:10.1007/978-1-62703-514-9_2
Conferences:
- Armando J. Pinho, Diogo Pratas, Paulo J. S. G. Ferreira. A new compressor for measuring distances among images. Proceedings of the International Conference on Image Analysis and Recognition, ICIAR 2014, Vilamoura, Portugal, vol. LNCS 8814, p. 30-37, October 2014. doi:10.1007/978-3-319-11758-4_4
- Diogo Pratas, Raquel M. Silva, Armando J. Pinho. Large-scale inversions between human reference assemblies. Proc. of the 20th Portuguese Conf. on Pattern Recognition, RecPad 2014, Covilha, Portugal, October 2014.
- Raquel M. Silva, Luisa Castro, Diogo Pratas, Armando J. Pinho. Towards personalized medicine: ebola virus absent words in the human genome. Proc. of the 20th Portuguese Conf. on Pattern Recognition, RecPad 2014, Covilha, Portugal, October 2014.
- Diogo Pratas, Armando J. Pinho. Exploring deep Markov models in genomic data compression using sequence pre-analysis. Proc. of the European Signal Processing Conference, EUSIPCO 2014, Lisbon, Portugal, September 2014.
- Diogo Pratas, Armando J. Pinho. A conditional compression distance that unveils insights of the genomic evolution. Data Compression Conference (DCC), 2014, vol., no., pp.421,421, 26-28 March 2014. doi:10.1109/DCC.2014.58
- Armando J. Pinho, Diogo Pratas, Paulo J S G Ferreira. Information Profiles for DNA Pattern Discovery. Data Compression Conference (DCC), 2014, vol., no., pp.420,420, 26-28 March 2014. doi:10.1109/DCC.2014.54
- Diogo Pratas, Armando J. Pinho. Insights into primates genomic evolution using a compression distance. Proc. of the 19th Portuguese Conf. on Pattern Recognition, RecPad 2013, Lisbon, Portugal, November 2013.
- Luis M. O. Matos, António J. R. Neves, Diogo Pratas, Armando J. Pinho. Lossless compression of symbolic and biomedical images using finite-context models. University of Aveiro Reseach Day 2013, Aveiro, Portugal, June 2013.
- Diogo Pratas, Armando J. Pinho. On the compression of FASTQ quality-scores. Proc. of the 18th Portuguese Conf. on Pattern Recognition, RecPad 2012, Coimbra, Portugal, October 2012.
- Sara Pinto Garcia, João M. O. S. Rodrigues, Diogo Pratas, Armando J. Pinho. Comparing maximal exact repeats in human genome assemblies using a normalized compression distance. 20th Annual International Conference on Intelligent Systems for Molecular Biology, Long Beach, California, USA, July 2012.
- Luís Matos, Diogo Pratas, Armando J. Pinho. Compression of Whole Genome Alignments Using a Mixture of Finite-Context Models. Proceedings of 9th International Conference on Image Analysis and Recognition, ICIAR 2012, Aveiro, Portugal, vol. Aurélio Campilho and Mohamed Kamel (Eds.): Part I, LNCS 7324, p. 359-366, June 2012.
- Diogo Pratas, Armando J. Pinho. On the Detection of Unknown Locally Repeating Patterns in Images. Proceedings of 9th International Conference on Image Analysis and Recognition, ICIAR 2012, Aveiro, Portugal, vol. Aurélio Campilho and Mohamed Kamel (Eds.): Part I, LNCS 7324, p. 158-165, June 2012. doi:10.1007/978-3-642-31295-3_19
- Diogo Pratas, Armando J. Pinho, Sara Pinto Garcia. Exon: A Web-Based Software Toolkit for DNA Sequence Analysis. Advances in Intelligent and Soft Computing, Proc. of the 6th Int. Conf. on Practical Applications of Computational Biology & Bioinformatics, PACBB 2012, Salamanca, Spain, vol. 154, p. 217-224, March 2012. doi:10.1007/978-3-642-28839-5_25
- Diogo Pratas, Armando J. Pinho, Sara Pinto Garcia. Computation of the normalized compression distance of DNA sequences using a mixture of finite-context models. Bioinformatics 2012: International Conference on Bioinformatics Models, Methods and Algorithms, Vilamoura, Portugal, February 2012.
- Armando J. Pinho, Diogo Pratas, Sara Pinto Garcia. Complexity Profiles of DNA Sequences Using Finite-Context Models. Information Quality in e-Health: 7th Conference of the Workgroup Human-Computer Interaction and Usability Engineering of the Austrian Computer Society, USAB 2011, Graz, Austria, vol. LNCS 7058, p. 75-82, November 2011.
- Diogo Pratas, Sara Pinto Garcia, Armando J. Pinho. Analysis of patterns in S. pombe genome through compression-based complexity profiles. Proc. of the 17th Portuguese Conf. on Pattern Recognition, RecPad 2011, Porto, Portugal, October 2011.
- Armando J. Pinho, Diogo Pratas, Paulo J S G Ferreira, Sara Pinto Garcia. Symbolic to numerical conversion of DNA sequences using finite-context models. Proc. of the European Signal Processing Conference, EUSIPCO 2011, Barcelona, Spain, August 2011.
- Diogo Pratas, Carlos A C Bastos, Armando J. Pinho, António J. R. Neves, Luís Matos. DNA synthetic sequences generation using multiple competing Markov models. Proc. of the IEEE Workshop on Statistical Signal Processing, SSP 2011, Nice, France, June 2011.
- Armando J. Pinho, Diogo Pratas, Paulo J S G Ferreira. Bacteria DNA sequence compression using a mixture of finite-context models. Proc. of the IEEE Workshop on Statistical Signal Processing, SSP 2011, Nice, France, June 2011.
- Diogo Pratas, Armando J. Pinho. Compressing the human genome using exclusively Markov models. Advances in Intelligent and Soft Computing, Proc. of the 5th Int. Conf. on Practical Applications of Computational Biology & Bioinformatics, PACBB 2011, Salamanca, Spain, vol. 93, p. 213-220, April 2011.
- Diogo Pratas, Armando J. Pinho, António J. R. Neves, Carlos A C Bastos. DNA synthetic sequences generated by finite-context models. Proc. of the 16th Portuguese Conf. on Pattern Recognition, RecPad 2010, October 2010.
- Diogo Pratas, Armando J. Pinho. Analysis of DNA sequences using finite-context modelling and compression. Proc. of the 16th Portuguese Conf. on Pattern Recognition, RecPad 2010, October 2010.
Thesis:
- Diogo Pratas. Compression and analysis of genomic data. Ph.D. Thesis, University of Aveiro, Aveiro, Portugal, January 2016.